Download presentation
Presentation is loading. Please wait.

1
Protein Sequencing KOREA UNIV. Chem. & Bio Eng. 박 기 태

2
Protein Amino acid Peptide Bonding 단백질은 아미노산들이 공유결합으로 연결된 중합체
Amino acid 단백질은 아미노산들이 공유결합으로 연결된 중합체 Side chain에 따라 종류가 달라짐 현재 22개의 종류가 발견 Amino acid Peptide Bonding Polypeptide 형성 일반적으로, amino acid가 50개 이하 : peptide amino acid가 50개 이상 : protein Dehydration Polypeptide

3
Joseph A. Krzycki Ph.D University of Wisconsin-Madison. Associate Professor Department of Microbiology Ohio State University (614) ;
 ;")
4
Amino Acids

5
Amino Acids (2)
")
6
Structures Primary Structure Secondary Structure Amino acid 의 연결순서
Backbone을 이루는 원자들의 규칙적이고 반복적인 배열에 의해 형성된 local structure

7
Structures (2) Tertiary Structure Quaternary Structure
Amino acid들이 공간상에서 특정하게 배열되는 것 (folding of peptide chain) 아미노산의 서열이 최종적인 삼차원 구조 결정 Tertiary structure에 의해 각 Protein의 고유기능 발현 Quaternary Structure Aggregation of two of more peptides
 아미노산의 서열이 최종적인 삼차원 구조 결정. Tertiary structure에 의해 각 Protein의 고유기능 발현. Quaternary Structure. Aggregation of two of more peptides.")
8
Combination n개의 amino acid로 이루어진 protein의 경우, 20개의 서로 다른 amino acid를 n개 배열하는 조합 = 20n 가지 100개의 amino acid로 구성된 protein의 경우, 가능한 조합은 가지로서 한가지 구조를 검색하는데 10-15초가 걸린다고 했을 때 가능한 모든 구조를 검색하는데 걸리는 시간은 약 1.27 X 10115초 ( 우주의 나이 : 약 1018초) 인체를 구성하는 단백질의 개수는 10만개로 추정되고 있으며, 구조가 밝혀진 것은 20%정도에 불과
 인체를 구성하는 단백질의 개수는 10만개로 추정되고 있으며, 구조가 밝혀진 것은 20%정도에 불과.")
10
Protein Sequence Analysis
실험적 방법 X-Ray Crystallography NMR Spectroscopy 예측적 방법 ab. initio Method Homology Modeling Theading

11
X-Ray Crystallography
물과 단백질로 이루어진 표본에서 물을 서서히 증발시켜 단결정 생성 결정의 원자를 둘러싸는 전자구름에 반사되는 X선 분석 단백질의 3차원 구조를 알 수 있는 가장 정확한 방법 수용성 단백질에만 적용 RU300, R-AXIS IV++ (Rigaku, Japan)
")
12
NMR Spectroscopy 두 수소원자 사이의 핵자기공명을 측정하여 nonzero spin을 가진 단백질 원자핵을 검출하여 모형화 결정을 만들 필요 없음 단백질이 물분자에 의해 완전히 둘러싸여 있어 세포내와 같은 환경에서 분석가능 Amino acid의 길이가 150개 이상인 단백질에는 적용이 어려움

13
ab. initio Method 기존의 단백질 구조 정보를 이용하지 않고 단백질을 구성하는 원자 수준에서 발생하는 물리 화학적 특성을 계산하여 구조 예측 Anfinsen의 열역학적 가설을 기반 단백질의 3차원구조는 주어진 조건에서 전체 계의 자유에너지가 가장 낮은 구조이다. 3차 구조는 아미노산의 서열(1차 구조)에 의해 결정된다. 단백질 분자의 원자간 상호작용 에너지, 전체 계의 에너지를 단백질 분자의 3차원 구조로 서술하여 그 최소값을 구함 Potential function의 예 V=1/2∑Kb(b-b0)2+1/2∑KΘ(Θ-Θ0) ∑KΦ(nΦ-δ)+∑(Aij/r6ij+ Bij/r12ij Cij/r10ij+ qiqj/Drij)
에 의해 결정된다. 단백질 분자의 원자간 상호작용 에너지, 전체 계의 에너지를 단백질 분자의 3차원 구조로 서술하여 그 최소값을 구함. Potential function의 예 V=1/2∑Kb(b-b0)2+1/2∑KΘ(Θ-Θ0)2 + ∑KΦ(nΦ-δ)+∑(Aij/r6ij+ Bij/r12ij + Cij/r10ij+ qiqj/Drij)")
14
Homology Modeling 서열이 비슷한 단백질은 구조와 기능도 비슷한 경우가 많다는 특성을 이용하여 기존의 알려진 단백질 Database(PDB)에서 의미 있는 서열상의 유사성을 찾아내는 과정 Query sequence – base sequence, amino acid sequence Algorithm – Smith Waterman, FASTA, BLAST

15
Smith-Waterman Dynamic Programming을 이용하여 임의의 서열과 데이터베이스에 저장된 서열들을 비교(PAM, BLUSUM) 비교적 정확한 검색 결과를 얻을 수 있지만 검색하는데 시간이 오래 걸린다. Scoring Matrix 비용(cast) 혹은 가중치(weight)의 개념 도입 W(a,a) =0 W(a,b) =1 W(a, -) = w(- ,b) =1 두 염기서열 비용계 1.AGCACAC-A AG-CACACA A-CACACTA ACACACT-A cost: cost:2 적절한 배열(optimal alignment) - 두 염기서열의 배열에 드는 비용이 최소한으로 드는 배열 문제를 해결하기 위해서 그 전 단계 문제의 답이 필요하고 다시 그 전 단계 문제의 답이 필요하여 Recursion으로 반복되는 경우, 가장 기본적인 문제의 답부터 BottomUp 방식으로 계산해 옴으로써 전체 문제를 해결하는 Algorithm. DivideAndConquer와 달리 중복된 계산을 하지 않으며, DivideAndConquer처럼 독립적인 부분문제로 나누어 접근하지 않고 종속적인 문제들의 일련을 순서에 맞게(순행 혹은 역행) 해결해 나간다. -- From DynamicProgramming BLOSUM (BLOks SUbstitution Matrix):1991년에 Altschul 등에 의해 발표된 BLOSUM은 현재 BLAST등의 검색에 제공되며 PAM과 함께 가장 많이 쓰이는 치환 행렬의 한 종류이다. BLOSUM은 Block database로부터 개발된 것으로, Block 데이터베이스는 아미노산 서열 중 다른 부분에 비해 굉장히 보존된 (conserved) 부분만을 모아 만든 데이터베이스이다. 이중 일부는 어떤 기능을 가진 motif로 알려져 있다. PAM이 연관된 서열들과 유추된 서열로부터 치환 확률을 구하는 반면 BLOSUM은 block내에서 아미노산들을 배열한 후 각각의 아미노산들이 짝(pair)을 이루는 확률을 관찰해서 치환 확률을 구한 것이다. 연속적인 치환 행렬을 만들기 위해 서열들을 각각의 block에 clustering을 시키고 clustering percentage는 각각의 group들에 포함시키기 위한 서열들의 최소한의 일치성 (identity)으로 정의한다. 예를 들면 clustering percentage가 35%라면 임의의 서열 A와 B를 배열시켰을 때 적어도 35% 이상의 identity를 가지고 있을 때 같은 group에 포함시키고 BLOSUM35로 정의한다. 또한 임의의 서열 C가 A와 B 둘 중 하나와 35 % 이상의 identity를 가질 경우에 또한 같은 group에 포함 시킨다. 각각의 배열된 아미노산 서열들의 pair들의 갯수를 센 후 서열 A,B,C가 각각 차지하는 비중을 평균하여 측정 행렬 값들을 구한다. 2. Gap penalties Gap penalty는 삽입 혹은 삭제에 의해 생기는 gap에 얼마의 감점 (penalty)를 줄 것인가를 정하는 것이다. 현재의 통계적 계산으로는 gap penalty를 얼마를 줄 것인가에 대한 정확한 해답은 없지만 여러가지 실험적 사실을 통해 -10, -2에서 -14, -4 정도가 적당하다고 한다. 첫 번째 값은 gap이 처음 생길 때 주는 감점이고, 두 번째 값은 그 다음에 생기는 연속적인 gap에 대한 감점이다. 예를 들면 두 개의 서열 사이에 4개의 gap이 있고, -10, -2의 값을 적용하면 전체 gap penalty는 -10+3×(-2) = -16이 된다. 이렇게 다른 값을 적용하는 이유는 진화상에서 처음 gap이 생기기는 힘들지만 그 이후 연속적으로 생기는 gap은 처음에 비해 쉽게 생길 수 있기 때문이다. 큰 gap penalty (예를 들면 -14, -4)는 partial sequence (EST 같은)의 비교에 적당하다. 사용자는 gap penalty를 조정함으로써 sensitivity를 조절 할 수 있다. 예를 들면 FASTA 검색에서 expectation value가 0.2 이하로 연관성이 거의 없는 서열들이 결과로 출력되었을 때 gap penalty의 값을 올림으로서 이런 서열들을 제거 해 나갈 수 있다.
 혹은 가중치(weight)의 개념 도입. W(a,a) =0. W(a,b) =1. W(a, -) = w(- ,b) =1. 두 염기서열 비용계. 1.AGCACAC-A 2. AG-CACACA. A-CACACTA ACACACT-A. cost:2 cost:2. 적절한 배열(optimal alignment) - 두 염기서열의 배열에 드는 비용이 최소한으로 드는 배열. 문제를 해결하기 위해서 그 전 단계 문제의 답이 필요하고 다시 그 전 단계 문제의 답이 필요하여 Recursion으로 반복되는 경우, 가장 기본적인 문제의 답부터 BottomUp 방식으로 계산해 옴으로써 전체 문제를 해결하는 Algorithm. DivideAndConquer와 달리 중복된 계산을 하지 않으며, DivideAndConquer처럼 독립적인 부분문제로 나누어 접근하지 않고 종속적인 문제들의 일련을 순서에 맞게(순행 혹은 역행) 해결해 나간다. -- From DynamicProgramming BLOSUM (BLOks SUbstitution Matrix):1991년에 Altschul 등에 의해 발표된 BLOSUM은 현재 BLAST등의 검색에 제공되며 PAM과 함께 가장 많이 쓰이는 치환 행렬의 한 종류이다. BLOSUM은 Block database로부터 개발된 것으로, Block 데이터베이스는 아미노산 서열 중 다른 부분에 비해 굉장히 보존된 (conserved) 부분만을 모아 만든 데이터베이스이다. 이중 일부는 어떤 기능을 가진 motif로 알려져 있다. PAM이 연관된 서열들과 유추된 서열로부터 치환 확률을 구하는 반면 BLOSUM은 block내에서 아미노산들을 배열한 후 각각의 아미노산들이 짝(pair)을 이루는 확률을 관찰해서 치환 확률을 구한 것이다. 연속적인 치환 행렬을 만들기 위해 서열들을 각각의 block에 clustering을 시키고 clustering percentage는 각각의 group들에 포함시키기 위한 서열들의 최소한의 일치성 (identity)으로 정의한다. 예를 들면 clustering percentage가 35%라면 임의의 서열 A와 B를 배열시켰을 때 적어도 35% 이상의 identity를 가지고 있을 때 같은 group에 포함시키고 BLOSUM35로 정의한다. 또한 임의의 서열 C가 A와 B 둘 중 하나와 35 % 이상의 identity를 가질 경우에 또한 같은 group에 포함 시킨다. 각각의 배열된 아미노산 서열들의 pair들의 갯수를 센 후 서열 A,B,C가 각각 차지하는 비중을 평균하여 측정 행렬 값들을 구한다. 2. Gap penalties. Gap penalty는 삽입 혹은 삭제에 의해 생기는 gap에 얼마의 감점 (penalty)를 줄 것인가를 정하는 것이다. 현재의 통계적 계산으로는 gap penalty를 얼마를 줄 것인가에 대한 정확한 해답은 없지만 여러가지 실험적 사실을 통해 -10, -2에서 -14, -4 정도가 적당하다고 한다. 첫 번째 값은 gap이 처음 생길 때 주는 감점이고, 두 번째 값은 그 다음에 생기는 연속적인 gap에 대한 감점이다. 예를 들면 두 개의 서열 사이에 4개의 gap이 있고, -10, -2의 값을 적용하면 전체 gap penalty는 -10+3×(-2) = -16이 된다. 이렇게 다른 값을 적용하는 이유는 진화상에서 처음 gap이 생기기는 힘들지만 그 이후 연속적으로 생기는 gap은 처음에 비해 쉽게 생길 수 있기 때문이다. 큰 gap penalty (예를 들면 -14, -4)는 partial sequence (EST 같은)의 비교에 적당하다. 사용자는 gap penalty를 조정함으로써 sensitivity를 조절 할 수 있다. 예를 들면 FASTA 검색에서 expectation value가 0.2 이하로 연관성이 거의 없는 서열들이 결과로 출력되었을 때 gap penalty의 값을 올림으로서 이런 서열들을 제거 해 나갈 수 있다.")
16
(예) s= AGCACAGA, t=ACACACTA 두 서열이 있을때 s와t를 각각행렬의
축으로 하고 unit cost model(일치=0,치환,삽입,삭제=1)을 이용하여 각각의 행렬의 항들을 채워나간다. (대각선은 일치, 혹은 치환을, 수평선은 삽입을, 수직선은 삭제를 나타낸다.) 대각선에 의해 표시된 행로에 의해 s,t를 배열하면 S = AGCACAC – A T = A - CACACTA
을 이용하여 각각의. 행렬의 항들을 채워나간다. (대각선은 일치, 혹은 치환을, 수평선은 삽입을, 수직선은 삭제를 나타낸다.) 대각선에 의해 표시된 행로에 의해 s,t를 배열하면. S = AGCACAC – A. T = A - CACACTA.")
17
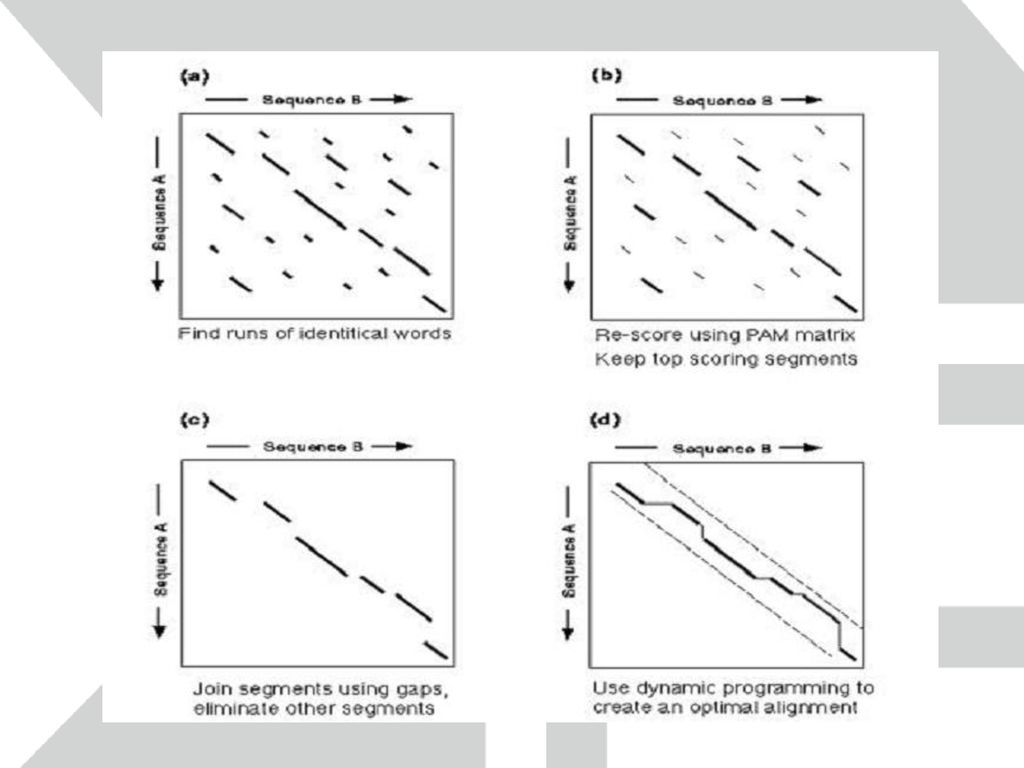
FASTA FASTA는 임의의 서열과 유사성을 가진 서열을 서열 데이터베이스로부터 찾는 프로그램
단백질 서열간의 비교를 위해 제작되었지만 염기 서열간의 비교도 가능 특히 TFASTA의 경우 입력한 단백질 서열과 염기 서열 데이터베이스 간의 비교도 가능 두 서열간의 dot blot을 그림으로서 비교를 시작 Dot blot에서 비슷한 서열을 가진 부분은 대각선으로 표시 문제를 해결하기 위해서 그 전 단계 문제의 답이 필요하고 다시 그 전 단계 문제의 답이 필요하여 Recursion으로 반복되는 경우, 가장 기본적인 문제의 답부터 BottomUp 방식으로 계산해 옴으로써 전체 문제를 해결하는 Algorithm. DivideAndConquer와 달리 중복된 계산을 하지 않으며, DivideAndConquer처럼 독립적인 부분문제로 나누어 접근하지 않고 종속적인 문제들의 일련을 순서에 맞게(순행 혹은 역행) 해결해 나간다. -- From DynamicProgramming BLOSUM (BLOks SUbstitution Matrix):1991년에 Altschul 등에 의해 발표된 BLOSUM은 현재 BLAST등의 검색에 제공되며 PAM과 함께 가장 많이 쓰이는 치환 행렬의 한 종류이다. BLOSUM은 Block database로부터 개발된 것으로, Block 데이터베이스는 아미노산 서열 중 다른 부분에 비해 굉장히 보존된 (conserved) 부분만을 모아 만든 데이터베이스이다. 이중 일부는 어떤 기능을 가진 motif로 알려져 있다. PAM이 연관된 서열들과 유추된 서열로부터 치환 확률을 구하는 반면 BLOSUM은 block내에서 아미노산들을 배열한 후 각각의 아미노산들이 짝(pair)을 이루는 확률을 관찰해서 치환 확률을 구한 것이다. 연속적인 치환 행렬을 만들기 위해 서열들을 각각의 block에 clustering을 시키고 clustering percentage는 각각의 group들에 포함시키기 위한 서열들의 최소한의 일치성 (identity)으로 정의한다. 예를 들면 clustering percentage가 35%라면 임의의 서열 A와 B를 배열시켰을 때 적어도 35% 이상의 identity를 가지고 있을 때 같은 group에 포함시키고 BLOSUM35로 정의한다. 또한 임의의 서열 C가 A와 B 둘 중 하나와 35 % 이상의 identity를 가질 경우에 또한 같은 group에 포함 시킨다. 각각의 배열된 아미노산 서열들의 pair들의 갯수를 센 후 서열 A,B,C가 각각 차지하는 비중을 평균하여 측정 행렬 값들을 구한다. 2. Gap penalties Gap penalty는 삽입 혹은 삭제에 의해 생기는 gap에 얼마의 감점 (penalty)를 줄 것인가를 정하는 것이다. 현재의 통계적 계산으로는 gap penalty를 얼마를 줄 것인가에 대한 정확한 해답은 없지만 여러가지 실험적 사실을 통해 -10, -2에서 -14, -4 정도가 적당하다고 한다. 첫 번째 값은 gap이 처음 생길 때 주는 감점이고, 두 번째 값은 그 다음에 생기는 연속적인 gap에 대한 감점이다. 예를 들면 두 개의 서열 사이에 4개의 gap이 있고, -10, -2의 값을 적용하면 전체 gap penalty는 -10+3×(-2) = -16이 된다. 이렇게 다른 값을 적용하는 이유는 진화상에서 처음 gap이 생기기는 힘들지만 그 이후 연속적으로 생기는 gap은 처음에 비해 쉽게 생길 수 있기 때문이다. 큰 gap penalty (예를 들면 -14, -4)는 partial sequence (EST 같은)의 비교에 적당하다. 사용자는 gap penalty를 조정함으로써 sensitivity를 조절 할 수 있다. 예를 들면 FASTA 검색에서 expectation value가 0.2 이하로 연관성이 거의 없는 서열들이 결과로 출력되었을 때 gap penalty의 값을 올림으로서 이런 서열들을 제거 해 나갈 수 있다. 그려진 대각선들의 합을 계산
 해결해 나간다. -- From DynamicProgramming BLOSUM (BLOks SUbstitution Matrix):1991년에 Altschul 등에 의해 발표된 BLOSUM은 현재 BLAST등의 검색에 제공되며 PAM과 함께 가장 많이 쓰이는 치환 행렬의 한 종류이다. BLOSUM은 Block database로부터 개발된 것으로, Block 데이터베이스는 아미노산 서열 중 다른 부분에 비해 굉장히 보존된 (conserved) 부분만을 모아 만든 데이터베이스이다. 이중 일부는 어떤 기능을 가진 motif로 알려져 있다. PAM이 연관된 서열들과 유추된 서열로부터 치환 확률을 구하는 반면 BLOSUM은 block내에서 아미노산들을 배열한 후 각각의 아미노산들이 짝(pair)을 이루는 확률을 관찰해서 치환 확률을 구한 것이다. 연속적인 치환 행렬을 만들기 위해 서열들을 각각의 block에 clustering을 시키고 clustering percentage는 각각의 group들에 포함시키기 위한 서열들의 최소한의 일치성 (identity)으로 정의한다. 예를 들면 clustering percentage가 35%라면 임의의 서열 A와 B를 배열시켰을 때 적어도 35% 이상의 identity를 가지고 있을 때 같은 group에 포함시키고 BLOSUM35로 정의한다. 또한 임의의 서열 C가 A와 B 둘 중 하나와 35 % 이상의 identity를 가질 경우에 또한 같은 group에 포함 시킨다. 각각의 배열된 아미노산 서열들의 pair들의 갯수를 센 후 서열 A,B,C가 각각 차지하는 비중을 평균하여 측정 행렬 값들을 구한다. 2. Gap penalties. Gap penalty는 삽입 혹은 삭제에 의해 생기는 gap에 얼마의 감점 (penalty)를 줄 것인가를 정하는 것이다. 현재의 통계적 계산으로는 gap penalty를 얼마를 줄 것인가에 대한 정확한 해답은 없지만 여러가지 실험적 사실을 통해 -10, -2에서 -14, -4 정도가 적당하다고 한다. 첫 번째 값은 gap이 처음 생길 때 주는 감점이고, 두 번째 값은 그 다음에 생기는 연속적인 gap에 대한 감점이다. 예를 들면 두 개의 서열 사이에 4개의 gap이 있고, -10, -2의 값을 적용하면 전체 gap penalty는 -10+3×(-2) = -16이 된다. 이렇게 다른 값을 적용하는 이유는 진화상에서 처음 gap이 생기기는 힘들지만 그 이후 연속적으로 생기는 gap은 처음에 비해 쉽게 생길 수 있기 때문이다. 큰 gap penalty (예를 들면 -14, -4)는 partial sequence (EST 같은)의 비교에 적당하다. 사용자는 gap penalty를 조정함으로써 sensitivity를 조절 할 수 있다. 예를 들면 FASTA 검색에서 expectation value가 0.2 이하로 연관성이 거의 없는 서열들이 결과로 출력되었을 때 gap penalty의 값을 올림으로서 이런 서열들을 제거 해 나갈 수 있다. 그려진 대각선들의 합을 계산.")
19
FASTA 염기 서열 혹은 단백질 서열간의 유사성 검사 TFASTA 입력한 단백질 서열과 데이터베이스의 염기 서열 translation 시킨 후 유사성 검사 LFASTA 두 단백질 혹은 염기 서열의 부분 유사성 검색(compare local similarity)을 수행한 후 부분 서열 배열(local sequence alignment)의 결과를 보여줌 PFASTA 두 서열의 부분 유사성 검색 후 부분 서열의 결과 를 그림으로 보여줌
을 수행한 후 부분 서열 배열(local sequence alignment)의 결과를 보여줌. PFASTA. 두 서열의 부분 유사성 검색 후 부분 서열의 결과 를 그림으로 보여줌.")
20
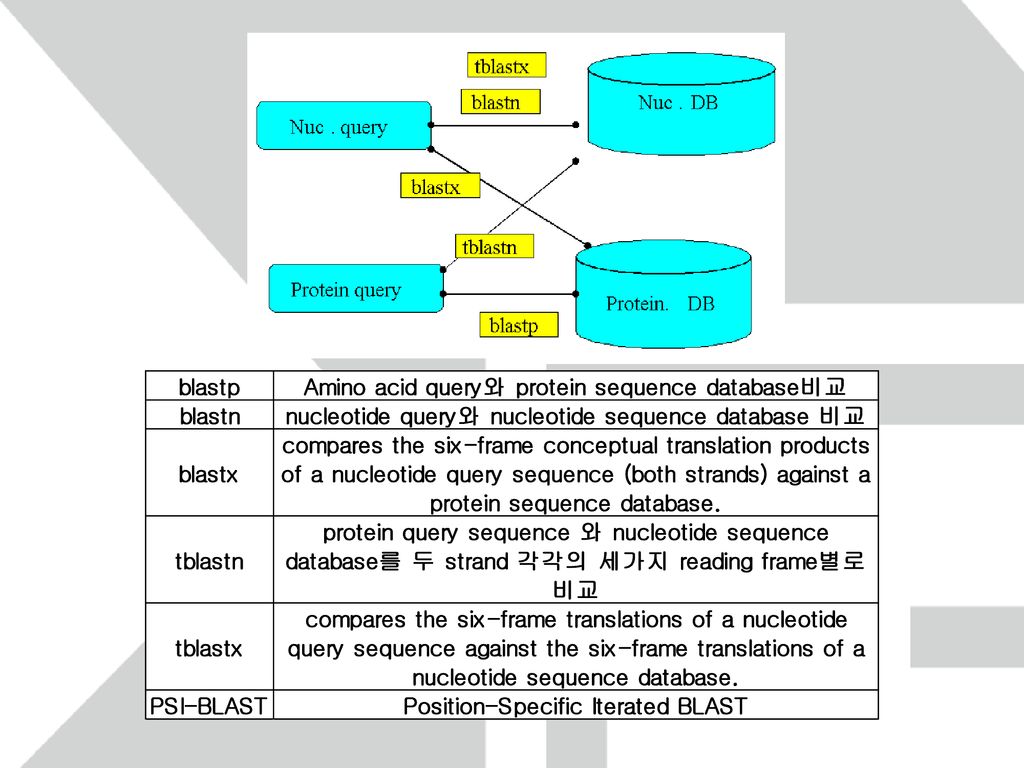
BLAST FASTA 의 속도를 높이기 위해 고안된 algorithm
Word length를 3(일반적으로 w로 표기)으로 하여 query에서 score가 특정 T보다 큰 words를 찾는다. Word list와 Database를 비교하여 일치하는 부분을 찾는다. 일치된 Word를 중심으로 양방향으로 확장하여 threadhold S 이상의 score를 가지는 alignment를 찾는다.
으로 하여 query에서 score가 특정 T보다 큰 words를 찾는다. Word list와 Database를 비교하여 일치하는 부분을 찾는다. 일치된 Word를 중심으로 양방향으로 확장하여 threadhold S 이상의 score를 가지는 alignment를 찾는다.")
23
Threading 대상서열을 이미 알려진 구조에 끼워 맞추어 가장 유사한 구조를 골라내는 방법
해당 원형(template)을 찾은 후에도 서열 구조를 맞추어야 한다. (protein folding 이용) 데이터베이스에서 유사성 있는 sequence를 찾고자 입력하는 sequence를 query sequence라고 합니다. 베이스 시퀀스의 경우 a t g c 의 4개의 문자로만 이루어져 있기 때문에 우연히 매칭이 될 확률이 높은 반면, 아미노산 시퀀스를 사용할 경우 20여개의 문자를 매칭하므로 좀더 엄밀한 매칭이 이루어 질수 있습니다. 시퀀싱에 사용되는 대표적인 알고리즘으로 Smith Waterman, FASTA, BLASTP가 있으며,
을 찾은 후에도 서열 구조를 맞추어야 한다. (protein folding 이용) 데이터베이스에서 유사성 있는 sequence를 찾고자 입력하는 sequence를 query sequence라고 합니다. 베이스 시퀀스의 경우 a t g c 의 4개의 문자로만 이루어져 있기 때문에 우연히 매칭이 될 확률이 높은 반면, 아미노산 시퀀스를 사용할 경우 20여개의 문자를 매칭하므로 좀더 엄밀한 매칭이 이루어 질수 있습니다. 시퀀싱에 사용되는 대표적인 알고리즘으로 Smith Waterman, FASTA, BLASTP가 있으며,")
24
Reference http://bric.postech.ac.kr/issue/aminoacid-05.html

Similar presentations



 정영림 (010-8797-6803, 기기분석학 Instrumental Analysis.>")











 컨설팅 사례연구ㅡ>")




.>")