Download presentation
Presentation is loading. Please wait.

1
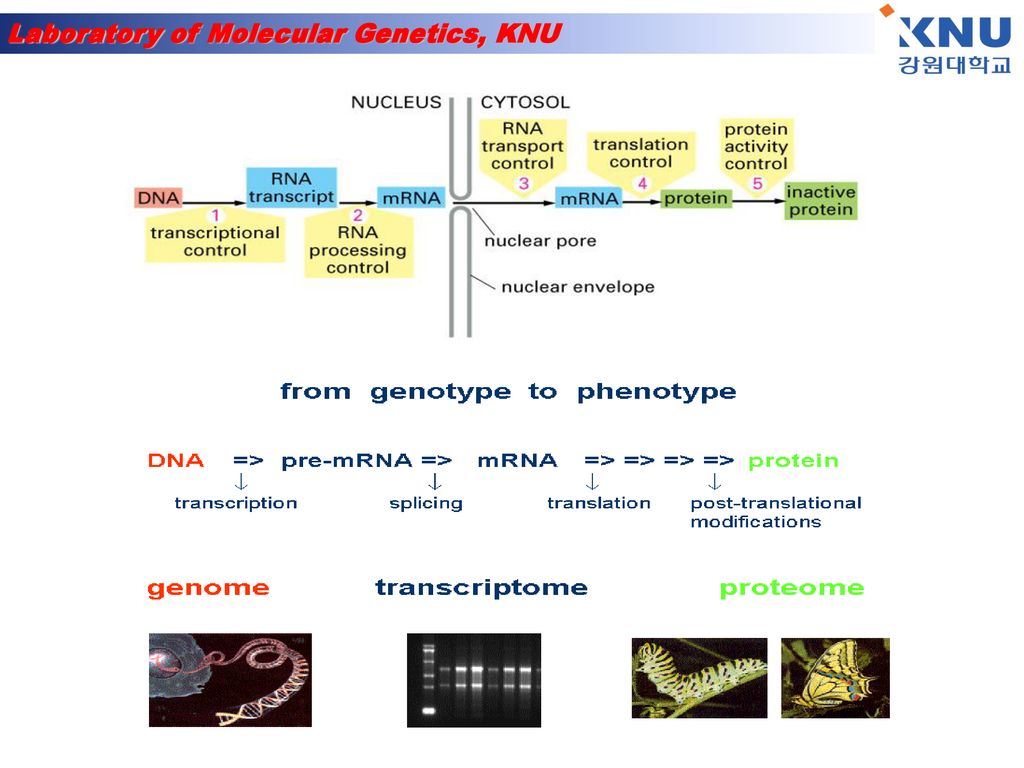
Gene expression & Analysis

2
Where are the genes located?
Genes are located on the chromosomes. Every species has a different number of chromosomes. There are two types of chromosomes: autosomes and sex chromosomes

3
Most of the time (90%) the genetic material in the form of chromatin.
Genes are located on the chromosomes which are found in the nucleus of a cell. When a cell is undergoing cell reproduction, the chromosomes are visible. Chromosomes appear when the chromatin condenses and become visible. Most of the time (90%) the genetic material in the form of chromatin. A genome is the complete genetic information contained in an individual. (gene + chromosome)
 the genetic material in the form of chromatin. A genome is the complete genetic information contained in an individual. (gene + chromosome)")
4
What is gene expression?
Gene expression is the activation of a gene that results in a protein.

5
Gene expression takes place differently in prokaryotes and eukaryotes.
No membrane bound organelles (nucleus) More primitive organisms Only one circular chromosome Bacteria are the only organisms that are prokaryotes. Eukaryotes Membrane bound organelles ( specialize in function –nucleus, mitochondria, chloroplast) Chromosomes are in pairs and not circular All organisms that are not bacteria: protist, fungi, plants and animals
 More primitive organisms. Only one circular chromosome. Bacteria are the only organisms that are prokaryotes. Eukaryotes. Membrane bound organelles ( specialize in function –nucleus, mitochondria, chloroplast) Chromosomes are in pairs and not circular. All organisms that are not bacteria: protist, fungi, plants and animals.")
6
DNA in eukaryotes has regions of coding and noncoding DNA
DNA in eukaryotes has regions of coding and noncoding DNA. The regions of DNA that code for proteins or traits are called EXONS, while the regions that do not code for proteins are called INTRONS. cytoplasm

7
In Eukaryotes, following mitosis or meiosis, DNA recoils but certain regions remain relaxed for transcription. The areas of relaxed DNA are called euchromatin. Transcription is the Reading of the DNA and Changing the code to mRNA. Translation is changing The mRNA into a trait by Using tRNA to interpret the

8
Translation RNA tRNA carries 3 base pair code for specific amino acid.
Single stranded Does not contain thymine but has uracil instead. tRNA carries 3 base pair code for specific amino acid. Amino acids compose polypeptid chains. One or more polypeptide chains compose a protein proteins provide the “blueprints” for our characteristics and functions.

10
Southern hybridization
핵산분리 제한효소처리 Agarose 전기영동 Gel 전처리 Southern blot Hybridization & detection

11
Southern Blotting 원리 Southern(1975)에 의해 개발된 방법으로, 그 순서를 요약하면 다음과 같다. - 해석할 DNA를 적당한 제한효소로 절단한다. - 절단된 DNA를 agarose gel 전기영동에 의해 분자량의 순으로 분획한다. - 변성된 DNA를 agarose gel로부터 nitrocellulose filter에 흡착시켜 고정시킨다. - 이렇게 준비된 filter와 방사선 동위원소로 표지한 probe를 수용액 중에서 hybridization - Filter를 X선 필름에 노출시켜 방사선으로 감광된 band를 검출함으로써 목적하는 DNA 의 존재를 확인하는 것이다.
에 의해 개발된 방법으로, 그 순서를 요약하면 다음과 같다. - 해석할 DNA를 적당한 제한효소로 절단한다. - 절단된 DNA를 agarose gel 전기영동에 의해 분자량의 순으로 분획한다. - 변성된 DNA를 agarose gel로부터 nitrocellulose filter에 흡착시켜 고정시킨다. - 이렇게 준비된 filter와 방사선 동위원소로 표지한 probe를 수용액 중에서 hybridization - Filter를 X선 필름에 노출시켜 방사선으로 감광된 band를 검출함으로써 목적하는 DNA 의 존재를 확인하는 것이다.")
16
Southern Blotting 실험방법
1. DNA의 절단 목적의 DNA를 적당한 제한효소로 절단한다. 2. Agarose gel 전기영동 Agarose gel은 두께 5 ~ 8mm 정도가 적당하며, 크기는 DNA분석용 gel보다 큰 것이 좋다. Agarose gel 전기영동은 보통 사용하는 방법과 같다. 3. DNA 단편을 Agarose gel로부터 filter에 흡착 DNA 단편들을 Agarose gel로부터 filter에 흡착하는 방법으로는 1) capillary transfer 2) Electrophoretic transfer 3) Vacuum transfer 등의 3가지가 있다. 여기에서는 가장 널리 쓰이는 capillary transfer에 대하여 설명한다. gel이 두꺼운 경우, 용액이 gel로 침루하는 시간이 다르기 때문에 두께 5 ~ 8mm 정도의 gel을 기준으로 한다. 4. Hybridization hybridization에 영향을 미치는 것은, 오노, 염농도, 시간, probe의 농도 등이다. 그러 나 특별한 경우를 제외하고는 온도는 65°C, 0.5 ~ 1.0M Nacl 농도에서 20시간 정도로 한다. Hybridization 방법은, 사용하는 probe의 종류(DNA, 합성 oligonucleotide, RNA) 따라 다르다, 그러므로 여기서는 가장 기본이 되는 DNA probe를 사용한 경우를 설 명하고자 한다. (1) 건조된 filter를 내열성 비닐포에 넣고 10mL의 prehybridization 용액을 가하여 적신 후, 기포를 제거하고 끝부분은 접착기로 봉한다. (2) 65°C, water bath에서 2 ~ 3시간 incubation한 후 비닐포의 한쪽 끝을 약간 절단하여 prehybridization 용액을 제거한다. (3) 5mL 정도의 hybridization용액 및 probe(25¡50ng)을 넣고, 기포를 제거한 후 다 시 비닐포의 한쪽 끝을 봉하여, 65 ~ 68°C에서 16 ~ 24 시간 incubation한다. (4) Hybridization이 끝난 filter를 적당한 용기에 넣어, 2X SSC-0.1% SDS로 65 ~ 68에서 10분간 천천히 흔들어 주면서 2회 세척한다. (5) 다시 1X SSC-0.1% SDS, 0.3X SSC-0.1% SDS, 0.1X SSC-0.1% SDS용액 순서 대로 65 ~ 68°C, 20분간 천천히 흔들어 주면서 세척한 후, Whatman 3 MM paper 위에 올려놓고 공기 중에서 건조시킨다. (6) Filter를 두장의 비닐랩 사이에 놓은후, 이것을 X-ray exposure cassette안에 놓 고, 접착 테이프를 이용하여 고정 시킨다. 5. Autoradiography 및 X-ray flim 현상 다음 (1), (3)단계는 암실, 안전등(RED FILTER)하에 실시한다. (1) 고정된 filter위에 X-ray film를 올려놓아 고정시킨후, 그 위에 intensifying screen 을 올려놓고 cassette 뚜껑을 덮는다. (2) 실온 또는 -70°C에서 2 ~ 3시간, 경우에 따라서는 1 ~ 2일 노출시킨다. (3) Film을 film holder에 걸어서 Kodak X-ray 현상액에서 2 ~ 3분 정도 담근다. (4) Film을 흐르는 물속에서 4 ~ 5분간 세척한다. (5) Kodak fixer액에 2 ~ 3분간 담근다. (6) Film을 흐르는 물속에서 10 ~ 30분 동안 세척한다. (7) Film을 걸어서 말린 후, 확인한다.
 capillary transfer 2) Electrophoretic transfer 3) Vacuum transfer 등의 3가지가 있다. 여기에서는 가장 널리 쓰이는 capillary transfer에 대하여 설명한다. gel이 두꺼운 경우, 용액이 gel로 침루하는 시간이 다르기 때문에 두께 5 ~ 8mm 정도의 gel을 기준으로 한다. 4. Hybridization hybridization에 영향을 미치는 것은, 오노, 염농도, 시간, probe의 농도 등이다. 그러 나 특별한 경우를 제외하고는 온도는 65°C, 0.5 ~ 1.0M Nacl 농도에서 20시간 정도로 한다. Hybridization 방법은, 사용하는 probe의 종류(DNA, 합성 oligonucleotide, RNA) 따라 다르다, 그러므로 여기서는 가장 기본이 되는 DNA probe를 사용한 경우를 설 명하고자 한다. (1) 건조된 filter를 내열성 비닐포에 넣고 10mL의 prehybridization 용액을 가하여 적신 후, 기포를 제거하고 끝부분은 접착기로 봉한다. (2) 65°C, water bath에서 2 ~ 3시간 incubation한 후 비닐포의 한쪽 끝을 약간 절단하여 prehybridization 용액을 제거한다. (3) 5mL 정도의 hybridization용액 및 probe(25¡50ng)을 넣고, 기포를 제거한 후 다 시 비닐포의 한쪽 끝을 봉하여, 65 ~ 68°C에서 16 ~ 24 시간 incubation한다. (4) Hybridization이 끝난 filter를 적당한 용기에 넣어, 2X SSC-0.1% SDS로 65 ~ 68에서 10분간 천천히 흔들어 주면서 2회 세척한다. (5) 다시 1X SSC-0.1% SDS, 0.3X SSC-0.1% SDS, 0.1X SSC-0.1% SDS용액 순서 대로 65 ~ 68°C, 20분간 천천히 흔들어 주면서 세척한 후, Whatman 3 MM paper 위에 올려놓고 공기 중에서 건조시킨다. (6) Filter를 두장의 비닐랩 사이에 놓은후, 이것을 X-ray exposure cassette안에 놓 고, 접착 테이프를 이용하여 고정 시킨다. 5. Autoradiography 및 X-ray flim 현상 다음 (1), (3)단계는 암실, 안전등(RED FILTER)하에 실시한다. (1) 고정된 filter위에 X-ray film를 올려놓아 고정시킨후, 그 위에 intensifying screen 을 올려놓고 cassette 뚜껑을 덮는다. (2) 실온 또는 -70°C에서 2 ~ 3시간, 경우에 따라서는 1 ~ 2일 노출시킨다. (3) Film을 film holder에 걸어서 Kodak X-ray 현상액에서 2 ~ 3분 정도 담근다. (4) Film을 흐르는 물속에서 4 ~ 5분간 세척한다. (5) Kodak fixer액에 2 ~ 3분간 담근다. (6) Film을 흐르는 물속에서 10 ~ 30분 동안 세척한다. (7) Film을 걸어서 말린 후, 확인한다.")
18
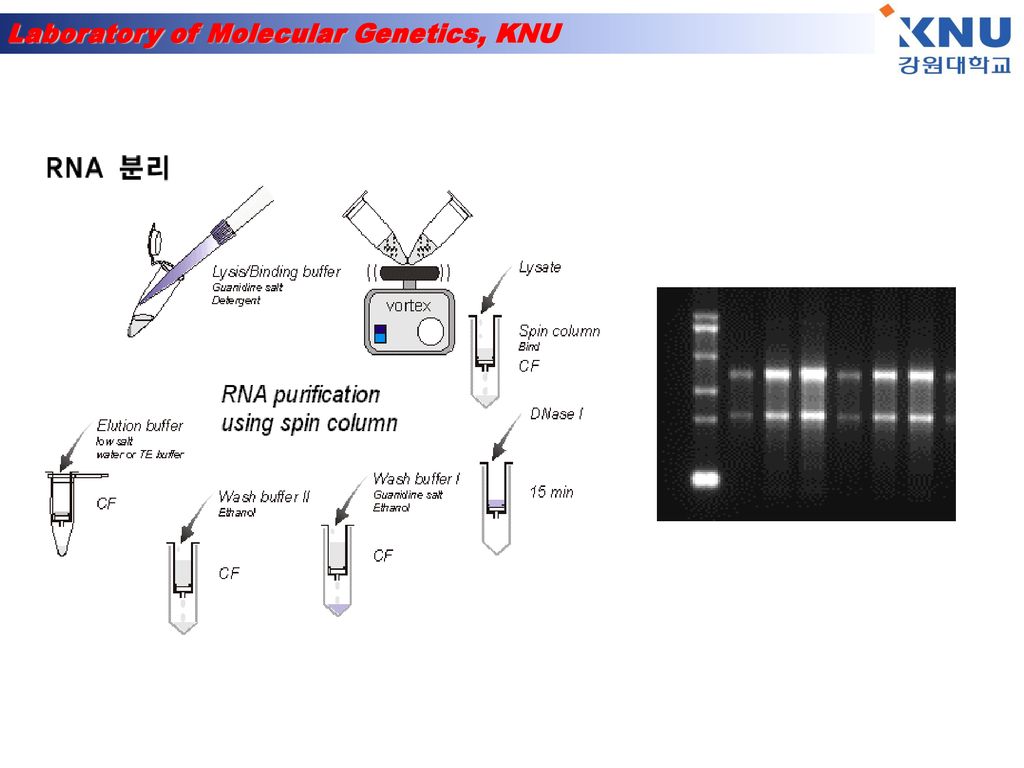
RNA 분리 1) Add 1㎖ TRIZOL 2) 5 min incubation at 15-30℃ 3) add 200㎕ chloroform. Shake tube vigorously by hand for 15 sec. 4) 2-3 min incubation at 15-30℃ 5) Centrifuge at 13,200×rpm for 15 min at 2-8℃ 6) Transfer upper aqueous phase 7) Add 500㎕ isopropyl alcohol (Precipitate the RNA) mix. 8) Incubate sample at 15-30℃ for 10 min 9) Centrifuge at 13,200×rpm for 10 min at 2-8℃→ pellet 없을 땐 1번 더 spin. 10) Remove supernatant(빠르게 제거) 11) Wash the RNA pellet once with 75% EtOH 1㎖(store 가능 : -20℃) 12) Mix the sample by vortexing and Centrifuge at 13,200×rpm 5min at 2-8℃ 13) Dry(air dry for 5min) 14) Add DEPC H2O 25㎕
 Add 1㎖ TRIZOL 2) 5 min incubation at 15-30℃ 3) add 200㎕ chloroform. Shake tube vigorously by hand for 15 sec. 4) 2-3 min incubation at 15-30℃ 5) Centrifuge at 13,200×rpm for 15 min at 2-8℃ 6) Transfer upper aqueous phase 7) Add 500㎕ isopropyl alcohol (Precipitate the RNA) mix. 8) Incubate sample at 15-30℃ for 10 min 9) Centrifuge at 13,200×rpm for 10 min at 2-8℃→ pellet 없을 땐 1번 더 spin. 10) Remove supernatant(빠르게 제거) 11) Wash the RNA pellet once with 75% EtOH 1㎖(store 가능 : -20℃) 12) Mix the sample by vortexing and Centrifuge at 13,200×rpm 5min at 2-8℃ 13) Dry(air dry for 5min) 14) Add DEPC H2O 25㎕")
21
Northern Blotting 원리 1975년 Southern EM이 제한효소를 절단해서 agarose gel 전기 영동으로 분획한 DNA 단편을 직접 Nitro-cellulose filter에 옮기는 방법을 개발했다. 이 Southern blot는 유전자 해석 수단으로서 아주 유용하기 때문에 그 후 널리 이용되고 있다. 1977년 Stark는 agarose gel 전기 영동으로 분획한 RNA를 filter에 옮기는 방법을 개발했다. 이 방법은 DNA에 대한Southern blot 와 대비시켜 Northern blot로 부르게 되었다. 기본적으로는 Southern blotting 과 동일하다. RNA를 전기 영동한 후 filter상에 고정하여 DNA(또는 RNA)를 probe로 하여 hybridization 해서 목적의 RNA를 검출한다. 그러나 RNA는 이차구조를 만들기 쉽기 때문에 gel 중에서 변성된 상태로 전기 영동을 시행한다. (1) 취급에 따른 일반적 주의 RNA에서 바른 정보를 얻기 위해서는 RNA를 가능한 한 분해되지 않은 상태로 조제하지 않으면 안된다. 특히 RNase에 의한 분해에 주의할 필요가 있다. RNase의 오염으로부터 시료를 지키기 위하여 고무장갑, disposable 제품, Autoclave, 건열 멸균, RNase 저해제 등을 이용한다. 유리기구, teflon제 기구는 세정후 증류수로 잘 헹구어서 건열한다. 전기 영동조 등 건열할 수 없는 기구는 세정 후 5%의 과산화수소수에 1시간 담그고 증류수로 잘 헹군다. 물은 Autoclave하여 사용한다. 시약은 autoclave 하든가 할 수 없는 것은 여과 멸균한다. 또 RNA는 DNA 와 달리 alkali에서 가수 분해되기 쉽다. 완충액 등의 pH가 alkali로 되지 않도록 주의 할 필요가 있다. (2 ) 추출조제 total RNA 나 poly(A)+RNA를 이용한다. RNA를 조제한 경우, 그 중 80% 정도가 rRNA이며 mRNA 중의 정보를 얻기 위해서는 가능한 한 rRNA의 혼입을 방지하는 것이 좋다. 목적의 mRNA가 적다고 생각되는 경우는 RNA를 Oligo(dT) column 처리를 한다. mRNA는 poly(A) 구조를 3`말단에 가지고 있기 때문에 이 조작으로 mRNA를 수 십배로 농축 정제가 가능하다. 그 외 단백질 혼입도 가능한 한피하지 않으면 안된다.
를 probe로 하여 hybridization 해서 목적의 RNA를 검출한다. 그러나 RNA는 이차구조를 만들기 쉽기 때문에 gel 중에서 변성된 상태로 전기 영동을 시행한다. (1) 취급에 따른 일반적 주의 RNA에서 바른 정보를 얻기 위해서는 RNA를 가능한 한 분해되지 않은 상태로 조제하지 않으면 안된다. 특히 RNase에 의한 분해에 주의할 필요가 있다. RNase의 오염으로부터 시료를 지키기 위하여 고무장갑, disposable 제품, Autoclave, 건열 멸균, RNase 저해제 등을 이용한다. 유리기구, teflon제 기구는 세정후 증류수로 잘 헹구어서 건열한다. 전기 영동조 등 건열할 수 없는 기구는 세정 후 5%의 과산화수소수에 1시간 담그고 증류수로 잘 헹군다. 물은 Autoclave하여 사용한다. 시약은 autoclave 하든가 할 수 없는 것은 여과 멸균한다. 또 RNA는 DNA 와 달리 alkali에서 가수 분해되기 쉽다. 완충액 등의 pH가 alkali로 되지 않도록 주의 할 필요가 있다. (2 ) 추출조제 total RNA 나 poly(A)+RNA를 이용한다. RNA를 조제한 경우, 그 중 80% 정도가 rRNA이며 mRNA 중의 정보를 얻기 위해서는 가능한 한 rRNA의 혼입을 방지하는 것이 좋다. 목적의 mRNA가 적다고 생각되는 경우는 RNA를 Oligo(dT) column 처리를 한다. mRNA는 poly(A) 구조를 3`말단에 가지고 있기 때문에 이 조작으로 mRNA를 수 십배로 농축 정제가 가능하다. 그 외 단백질 혼입도 가능한 한피하지 않으면 안된다.")
22
Northern Blotting 실험방법
1. agarose gel의 제작(1% agarose, 1 x MOPS, 2.2mol/l formaldehyde) 2. RNA sample의 조제(RT-PCR RNA 준비과정과 동일 ) 3. 전기영동(UV 조사) V, 2 - 3시간(2 - 3분간) 4. gel을 꺼내고 10 x SSC에 담근다 - 20분 - 1시간 (NaCl, sodium citrate) 5. transfer - 하룻밤 overnight 6. 건열 처리 - 80℃, 30분 - 2시간 7. prehybridization - 42℃, 2 - 3시간 8. hybridization - 42℃, 하룻밤 overnight 9. 세 정 - 1 x SSC, 0.1% SDS, 실온, 20분간 1회 0.2 x SSC, 0.1% SDS, 68℃, 20분간 3회 10. autoradiography Northern Blotting 실험방법
 2. RNA sample의 조제(RT-PCR RNA 준비과정과 동일 ) 3. 전기영동(UV 조사) V, 2 - 3시간(2 - 3분간) 4. gel을 꺼내고 10 x SSC에 담근다 - 20분 - 1시간 (NaCl, sodium citrate) 5. transfer - 하룻밤 overnight 6. 건열 처리 - 80℃, 30분 - 2시간 7. prehybridization - 42℃, 2 - 3시간 8. hybridization - 42℃, 하룻밤 overnight 9. 세 정 - 1 x SSC, 0.1% SDS, 실온, 20분간 1회 0.2 x SSC, 0.1% SDS, 68℃, 20분간 3회 10. autoradiography. Northern Blotting 실험방법.")
23
Detection of alternative splicing by Northern blotting
Northern blotting can be used to detect specific RNAs in complex mixtures. Southern blotting detects specific DNA fragments. Western blotting (immunoblotting) detects specific proteins with antibodies. RNA mixture Transfer solution RNA Question: You are using Northern blotting to analyze two mRNA samples derived from fibroblasts and hepatocytes. What will you see if you use a probe made from exon EIIIB of the fibronectin gene? What about using a probe made from the exon next to EIIIB?
 detects specific proteins with antibodies. RNA. mixture. Transfer solution. RNA. Question: You are using Northern blotting to analyze two mRNA samples derived from fibroblasts and hepatocytes. What will you see if you use a probe made from exon EIIIB of the fibronectin gene What about using a probe made from the exon next to EIIIB")
24
RT-PCR 과정 ■ RT-PCR은 세가지 과정
(1) RNA 분리 과정 (이 과정은 Northern Blot을 하기 전에 시행해야 하는 동일한 과정이다) (2) cDNA 합성 과정(reverse transcription) (3) PCR amplification (이 과정은 Genomic DNA로부터 특정 유전자 부위를 증폭시키는 과정과 같다) 위의 세가지 과정으로 진행된다. mRNA로부터 reverse transcriptase를 이용하여 cDNA를 제조하는 방법에는 어떤 oligonucleotide를 primer로 사용하는가에 따라 세가지 방법 즉, (1) Antisense primer(3' 쪽 유전자에 특이성을 지닌 primer)를 이용하여 특정 부위 cDNA 제조 (2) Random hexamer를 이용하여 전체 mRNA에 상보적인 cDNA 제조 (3) Oligo dT primer를 이용하여 전체 mRNA에 상보적인 cDNA 제조가 있다. 여기서는 oligo dT primer를 이용한 방법을 사용하였다. 또한 Northern Blot과 마찬가지로 cDNA가 만들어질 때까지는 RNase에 의한 오염 방지에 신경을 써야 한다.
 RNA 분리 과정 (이 과정은 Northern Blot을 하기 전에 시행해야 하는 동일한 과정이다) (2) cDNA 합성 과정(reverse transcription) (3) PCR amplification (이 과정은 Genomic DNA로부터 특정 유전자 부위를 증폭시키는 과정과 같다) 위의 세가지 과정으로 진행된다. mRNA로부터 reverse transcriptase를 이용하여 cDNA를 제조하는 방법에는 어떤 oligonucleotide를 primer로 사용하는가에 따라 세가지 방법 즉, (1) Antisense primer(3 쪽 유전자에 특이성을 지닌 primer)를 이용하여 특정 부위 cDNA 제조 (2) Random hexamer를 이용하여 전체 mRNA에 상보적인 cDNA 제조 (3) Oligo dT primer를 이용하여 전체 mRNA에 상보적인 cDNA 제조가 있다. 여기서는 oligo dT primer를 이용한 방법을 사용하였다. 또한 Northern Blot과 마찬가지로 cDNA가 만들어질 때까지는 RNase에 의한 오염 방지에 신경을 써야 한다.")
25
RT-PCR 원리 RT-PCR이란 P.Seeburg(1986)에 의해 RNA를 찾고 분석하는데 도입된 방법으로 mRNA(messenger RNA)로부터 reverse transcription 과정을 통해 얻어진 cDNA(complementary DNA)를 PCR로 증폭하는 방법이다. 이러한 방법은 RNA 검사의 sensitivity를 높이고 소량의 RNA로부터 염기서열을 분석할 수 있게 하였다. 맨 먼저 reverse transcriptase로 RNA를 complementary DNA(cDNA)로 역전사합니다. 이때 쓰이는 primer는 downstream primer뿐입니다. 왜냐하면 RNA는 single strand이기 때문입니다. 그후 생성된 cDNA에서 일반적인 PCR의 과정을 밟아서 유전자를 증폭합니다.
에 의해 RNA를 찾고 분석하는데 도입된 방법으로 mRNA(messenger RNA)로부터 reverse transcription 과정을 통해 얻어진 cDNA(complementary DNA)를 PCR로 증폭하는 방법이다. 이러한 방법은 RNA 검사의 sensitivity를 높이고 소량의 RNA로부터 염기서열을 분석할 수 있게 하였다. 맨 먼저 reverse transcriptase로 RNA를 complementary DNA(cDNA)로 역전사합니다. 이때 쓰이는 primer는 downstream primer뿐입니다. 왜냐하면 RNA는 single strand이기 때문입니다. 그후 생성된 cDNA에서 일반적인 PCR의 과정을 밟아서 유전자를 증폭합니다.")
26
그러면 downstream primer는 어떻게 만들까요. 크게 다음과 같은 세가지 방법이 알려져 있습니다
그러면 downstream primer는 어떻게 만들까요. 크게 다음과 같은 세가지 방법이 알려져 있습니다. mRNA 뒤에 poly(A) tail이 붙는 것을 응용한 oligo(dT) primer, gene specific primer, 그리고 여기저기 붙는 random primer입니다. Oligo(dT) primer를 쓰면 분리한 RNA 중 mRNA는 모두 cDNA로 만들어집니다. Gene specific primter를 사용하면 target gene만 cDNA로 만들어지겠지요. 그리고 random primer(주로 hexamer)는 염기서열이 일정치 않지만 길이가 고정된 primer입니다. 이런 primer를 넣고 반응시키면 길이도 다양하고 모든 RNA(rRNA, tRNA 포함)가 모두 cDNA로 만들어지겠지요.
 tail이 붙는 것을 응용한 oligo(dT) primer, gene specific primer, 그리고 여기저기 붙는 random primer입니다. Oligo(dT) primer를 쓰면 분리한 RNA 중 mRNA는 모두 cDNA로 만들어집니다. Gene specific primter를 사용하면 target gene만 cDNA로 만들어지겠지요. 그리고 random primer(주로 hexamer)는 염기서열이 일정치 않지만 길이가 고정된 primer입니다. 이런 primer를 넣고 반응시키면 길이도 다양하고 모든 RNA(rRNA, tRNA 포함)가 모두 cDNA로 만들어지겠지요.")
28
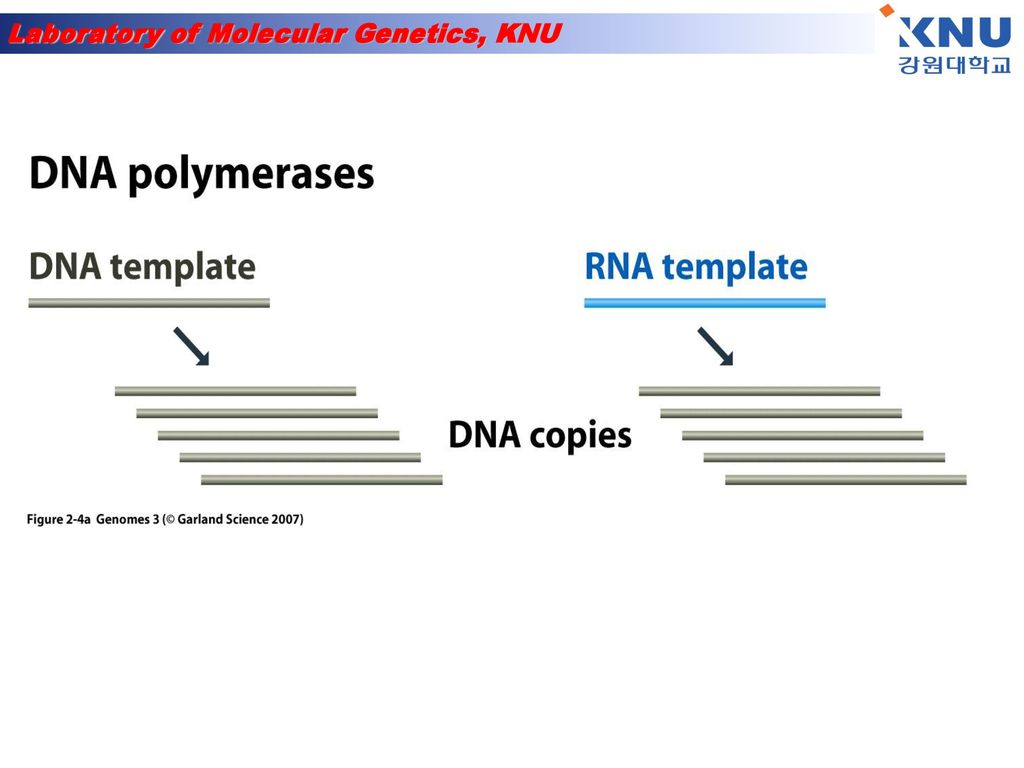
DNA synthesis is done by an enzyme (DNA polymerase) adding nucleotides to the 3’-end of a primer DNA chain
 adding nucleotides to the 3’-end of a primer DNA chain")
29
Polymerase Chain Reaction (PCR)-1
A pre-defined DNA sequence in the genome can be greatly amplified by repeated Polymerization cycles using 2 primers which hybridize to the ends of the target DNA. In each cycle, the amount of target DNA is doubled. After 10, 20 and 30 cycles, there is a 1000-, million- and billion-fold amplification respectively.

30
Polymerase Chain Reaction (PCR)-2
Each PCR cycle has 3 steps- Melting of DNA b. Hybridization of primer c. DNA synthesis

31
Nitrocellulose membrane
Western blot Antigen- Antibody reaction을 이용하여 여러 단백질 혼합물 중에서 원하는 특정 단백질만을 찾아내는 방법 Secondary antibody Primary antibody Block solution Nitrocellulose membrane antigen

32
Western Blotting 원리

33
Polyacrylamide Gel Electrophoresis:
From Gel to Blot Polyacrylamide Gel Electrophoresis: Break protein complexes into individual proteins Separates protein samples based on size Western Blot Analysis: Transfer the proteins to a nitrocellulose membrane More stable and permanent Identifies proteins by immunodetection: using specific antibodies against the protein of interest

34
Mini Trans-Blot Transfer Cell

35
Prepare for Electrophoretic Transfer
Place the closed and locked cassette in the electrode module Add the frozen Bio-Ice cooling unit and place in tank Fill the tank with buffer A stir bar can be added to help maintain the ion and temperature distribution in the tank even

36
Transfer Proteins from the gel to the nitrocellulose membrane minutes 100V Blotting buffer 1x Tris glycine with 20% ethanol Electric Current

37
Add the Primary Antibody anti- myosin light-chain Wash
Discard blocking solution Pour 10ml of primary antibody onto the membrane and gently rock for 10 minutes Primary antibody will bind to the myosin light-chain Quickly rinse membrane in 50ml of wash buffer and discard the wash buffer Add 50ml of wash leave for 3 minutes on the rocking platform Add the Primary Antibody anti- myosin light-chain Wash

38
Add Enzyme-linked Secondary Antibody Wash
Discard wash solution Pour 10ml of the secondary antibody onto the membrane and gently rock for 10 minutes Secondary antibody will bind to the primary antibody Quickly rinse membrane in 50ml of wash buffer and discard the wash buffer Add 50ml of wash leave for 3 minutes on the rocking platform Western Blot animation Add Enzyme-linked Secondary Antibody Wash

39
Western Blotting 실험과정 A. SAMPLE PREPARATION
Sample preparation procedures are provided for monolayer cells, suspension cells, and tissue samples. Follow the procedure suited to your needs. Monolayer Cells • Grow cells to subconfluency in a 100 mm x 20 mm petri dish, remove culture medium and rinse cell monolayer with room temperature 1x PBS. The following steps should be performed on ice or at 4° C using fresh, ice cold buffers. Add 0.6 ml of RIPA buffer to the monolayer cells in the plate. Gently rock plate for 15 minutes at 4° C. Remove adherent cells with a cell scraper. Transfer the resulting lysate to a microcentrifuge tube. Wash the plate once with 0.3 ml of RIPA buffer and combine with first lysate. (Optional: Add 10 µl of 10 mg/ml PMSF and/or pass through a 21-gauge needle to shear the DNA.) Incubate 30–60 minutes on ice. Centrifuge cell lysate at 10,000xg for 10 minutes at 4° C. The supernatant fluid is the total cell lysate. Transfer the supernatant to a new microcentrifuge tube. This is your whole cell lysate. For increased protein recovery, resuspend the pellet in a small volume of RIPA, centrifuge and combine supernantants. Suspension Cells • Collect approximately 2.0 x 107 cells by low-speed centrifugation (e.g. 200xg) at room temperature for 5 minutes. Carefully remove culture medium. Wash the pellet with PBS at room temperature, and again collect by low-speed centrifugation. Carefully remove supernatant. Add 1.0 ml of ice cold RIPA buffer with freshly added (Protease Inhibitors) and/or (Phosphatase Inhibitors). Gently resuspend cells in RIPA buffer with a pipet and incubate on ice for 30 minutes. Further disrupt and homogenize cells by hydrodynamic shearing (21-gauge needle), dounce homogenization or sonication, taking care not to raise the temperature of the lysate. (Optional: Add 10 µl of 10 mg/ml PMSF) Incubate 30 minutes on ice. Transfer to microcentrifuge tube(s) and centrifuge at 10,000xg for 10 minutes at 4° C. The supernatant fluid is the total cell lysate. Transfer the supernatant to a new microfuge tube. This is your whole cell lysate. For increased protein recovery, resuspend the pellet in a small volume of RIPA, centrifuge and combine supernantants.
 Incubate 30–60 minutes on ice. Centrifuge cell lysate at 10,000xg for 10 minutes at 4° C. The supernatant fluid is the total cell lysate. Transfer the supernatant to a new microcentrifuge tube. This is your whole cell lysate. For increased protein recovery, resuspend the pellet in a small volume of RIPA, centrifuge and combine supernantants. Suspension Cells. • Collect approximately 2.0 x 107 cells by low-speed centrifugation (e.g. 200xg) at room temperature for 5 minutes. Carefully remove culture medium. Wash the pellet with PBS at room temperature, and again collect by low-speed centrifugation. Carefully remove supernatant. Add 1.0 ml of ice cold RIPA buffer with freshly added (Protease Inhibitors) and/or (Phosphatase Inhibitors). Gently resuspend cells in RIPA buffer with a pipet and incubate on ice for 30 minutes. Further disrupt and homogenize cells by hydrodynamic shearing (21-gauge needle), dounce homogenization or sonication, taking care not to raise the temperature of the lysate. (Optional: Add 10 µl of 10 mg/ml PMSF) Incubate 30 minutes on ice. Transfer to microcentrifuge tube(s) and centrifuge at 10,000xg for 10 minutes at 4° C. The supernatant fluid is the total cell lysate. Transfer the supernatant to a new microfuge tube. This is your whole cell lysate. For increased protein recovery, resuspend the pellet in a small volume of RIPA, centrifuge and combine supernantants.")
40
B. ELECTROPHORESIS • Mix sample (40–60 µg whole cell lysate, 10–20 µg nuclear extract or 10–20 ng purified protein per lane) with an equal volume of 2x electrophoresis sample buffer and boil for 2–3 minutes. Unused samples may be stored at -20° C. Load up to 10 µl of lysate per 1.0 mm of well width for gels of 0.75 mm thickness. We recommend the use of Cruz Marker™ molecular weight standards. Load 2 µl/well for 0.75 mm gels and 5 µl/well for 1.5 mm gels. When used with Cruz Marker™ compatible secondary antibodies, internal standard bands will appear when the probed blot is exposed to detection reagent. Alternatively, use Prestained Molecular Weight Standards. Electrophorese according to standard protocols. Transfer proteins from the gel to a nitrocellulose or PVDF membrane using an electroblotting apparatus according to the manufacturer´s protocols. C. IMMUNOBLOTTING • Block non-specific binding by incubating membrane in Blotto (either Blotto A or Blotto B; IgG-free BSA, is recommended when using anti-bovine secondary antibodies) for 30–60 minutes at room temperature. Alternatively, the membrane may be blocked at 4° C overnight in a covered container, using Blotto without Tween-20. If using a phospho-specific antibody, add 0.01% (v/v) of each Phosphatase Inhibitor Cocktails to the blocking solution and the antibody diluent to inhibit phosphatases. Incubate the blocked membrane in primary antibody diluted in Blotto for 1 hour at room temperature. (For phospho-specific antibodies: Use Blotto B with 0.01% (v/v) of each Phosphatase Inhibitor Cocktails Optimal antibody concentration should be determined by titration. We recommend a starting dilution of µg/ml. Wash membrane three times for 5 minutes each with TBST. Incubate the membrane for 45 minutes at room temperature with horseradish peroxidase (HRP) conjugated secondary antibody, or alkaline phosphatase (AP) conjugated secondary antibody (Conventional Secondary Antibodies for Western Blotting), diluted to 1:500–1:2000 in Blotto. If high backgrounds are observed, secondary antibody should be diluted further (up to 1:20,000). If Cruz Marker™ molecular weight standards are used in the gel, the Cruz Marker™ compatible secondary antibodies must be used in order to visualize standards with ECL. Wash membrane three times for 5 minutes each with TBST and once for 5 minutes with TBST. Incubate membrane in Chemiluminescence Luminol Reagent according to Luminol data sheet, or visualize proteins using standard protocols. If luminol is used for visualization, an HRP-conjugated secondary antibody must be used.
 with an equal volume of 2x electrophoresis sample buffer and boil for 2–3 minutes. Unused samples may be stored at -20° C. Load up to 10 µl of lysate per 1.0 mm of well width for gels of 0.75 mm thickness. We recommend the use of Cruz Marker™ molecular weight standards. Load 2 µl/well for 0.75 mm gels and 5 µl/well for 1.5 mm gels. When used with Cruz Marker™ compatible secondary antibodies, internal standard bands will appear when the probed blot is exposed to detection reagent. Alternatively, use Prestained Molecular Weight Standards. Electrophorese according to standard protocols. Transfer proteins from the gel to a nitrocellulose or PVDF membrane using an electroblotting apparatus according to the manufacturer´s protocols. C. IMMUNOBLOTTING. • Block non-specific binding by incubating membrane in Blotto (either Blotto A or Blotto B; IgG-free BSA, is recommended when using anti-bovine secondary antibodies) for 30–60 minutes at room temperature. Alternatively, the membrane may be blocked at 4° C overnight in a covered container, using Blotto without Tween-20. If using a phospho-specific antibody, add 0.01% (v/v) of each Phosphatase Inhibitor Cocktails to the blocking solution and the antibody diluent to inhibit phosphatases. Incubate the blocked membrane in primary antibody diluted in Blotto for 1 hour at room temperature. (For phospho-specific antibodies: Use Blotto B with 0.01% (v/v) of each Phosphatase Inhibitor Cocktails Optimal antibody concentration should be determined by titration. We recommend a starting dilution of µg/ml. Wash membrane three times for 5 minutes each with TBST. Incubate the membrane for 45 minutes at room temperature with horseradish peroxidase (HRP) conjugated secondary antibody, or alkaline phosphatase (AP) conjugated secondary antibody (Conventional Secondary Antibodies for Western Blotting), diluted to 1:500–1:2000 in Blotto. If high backgrounds are observed, secondary antibody should be diluted further (up to 1:20,000). If Cruz Marker™ molecular weight standards are used in the gel, the Cruz Marker™ compatible secondary antibodies must be used in order to visualize standards with ECL. Wash membrane three times for 5 minutes each with TBST and once for 5 minutes with TBST. Incubate membrane in Chemiluminescence Luminol Reagent according to Luminol data sheet, or visualize proteins using standard protocols. If luminol is used for visualization, an HRP-conjugated secondary antibody must be used.")
41
Transfection and Protein localization

42
Exploring protein function
1) Where is it localized in the cell? Approaches: a) Make antibodies - immunofluorescence b) “Express” the protein in cells with a tag Fuse to GFP 2) What is it doing in the cell? Approaches: a) Reduce protein levels - RNA interference b) Increase protein levels “over-express” c) “Express” mutant versions Transfection!!!!
 Where is it localized in the cell Approaches: a) Make antibodies - immunofluorescence. b) Express the protein in cells with a tag. Fuse to GFP. 2) What is it doing in the cell Approaches: a) Reduce protein levels - RNA interference. b) Increase protein levels over-express c) Express mutant versions. Transfection!!!!")
43
Introduction of DNA into mammalian cells
Transfection = Introduction of DNA into mammalian cells Gene is transcribed and translated into protein = “expressed”

44
Direct introduction of the DNA
Electroporation - electric field temporarily disrupts plasma membrane Biolistics (gene gun)- fire DNA coated particles into cell Microinjection
- fire DNA coated particles into cell. Microinjection.")
45
Virally-mediated introduction of the DNA
Infection: Use recombinant viruses to deliver DNA Retroviruses Adenoviruses

46
Carrier-mediated introduction of the DNA
Positively charged carrier molecules are mixed with the DNA and added to cell culture media: Calcium Phosphate DEAE Dextran liposomes micelles Carrier-DNA complexes bind to plasma membrane and are taken up

47
Types of Transfection Transient:
Expression assayed hours post transfection Stable: Integration of the transfected DNA into the cell genome - selectable marker like neomycin resistance required “stably transfected” cell line

48
DNA “expression” vector transfected:
Insert gene in here For expression in cells Polyadenylation site GFP CMV Promoter Promoter SV40 To generate stable cell line pCMV/GFP resistance Ampicillin resistance Neomycin For amplification of the plasmid in bacteria Polyadenylation site pUC Bacterial origin of replication

49
Three ways to make Green fluorescent protein “GFP” fusion constructs:
PROTEIN X GFP GFP PROTEIN Y PROTEIN GFP Z

50
EXPERIMENT: Transfect unknown GFP fusion protein Protein X, Y or Z
Visualize GFP protein fluorescence by fluorescence microscopy in living cells Counter-stain with known marker to compare localization patterns in living cells = “vital stain”

51
Transfection 원리

52
Transfection 실험방법 Procedure
1. The day before transfection, seed 2–8 x 105 cells per 60 mm dish in 5 ml appropriate growth medium. The cell number seeded should produce 40–80% confluence on the day of transfection. 2. Incubate the cells under their normal growth conditions (generally 37°C and 5% CO2). 3. On the day of transfection, dilute 5 µg DNA with cell growth medium containing no serum, proteins, or antibiotics to a total volume of 150 µl. (For primary cells, use 2.5 µg plasmid DNA). Mix and spin down the solution for a few seconds to remove drops from the top of the tube. 4. Add 30 µl SuperFect Transfection Reagent to the DNA solution. (For primary cells, use 15 µl SuperFect Reagent). Mix by pipetting up and down 5 times, or by vortexing for 10 s. 5. Incubate the samples for 5–10 min at room temperature (15–25°C) to allow transfection-complex formation. 6. While complex formation takes place, gently aspirate the growth medium from the dish, and wash cells once with 4 ml PBS (phosphate buffer). 7. Add 1 ml cell growth medium (containing serum and antibiotics) to the reaction tube containing the transfection complexes. Mix by pipetting up and down twice, and immediately transfer the total volume to the cells in the 60 mm dishes. 8. Incubate cells with the transfection complexes for 2–3 h under their normal growth conditions. 9. Remove medium containing the remaining complexes from the cells by gentle aspiration, and wash cells once with 4 ml PBS. 10. Add fresh cell growth medium (containing serum and antibiotics). Assay cells for expression of the transfected gene after an appropriate incubation time.
. 3. On the day of transfection, dilute 5 µg DNA with cell growth medium containing no serum, proteins, or antibiotics to a total volume of 150 µl. (For primary cells, use 2.5 µg plasmid DNA). Mix and spin down the solution for a few seconds to remove drops from the top of the tube. 4. Add 30 µl SuperFect Transfection Reagent to the DNA solution. (For primary cells, use 15 µl SuperFect Reagent). Mix by pipetting up and down 5 times, or by vortexing for 10 s. 5. Incubate the samples for 5–10 min at room temperature (15–25°C) to allow transfection-complex formation. 6. While complex formation takes place, gently aspirate the growth medium from the dish, and wash cells once with 4 ml PBS (phosphate buffer). 7. Add 1 ml cell growth medium (containing serum and antibiotics) to the reaction tube containing the transfection complexes. Mix by pipetting up and down twice, and immediately transfer the total volume to the cells in the 60 mm dishes. 8. Incubate cells with the transfection complexes for 2–3 h under their normal growth conditions. 9. Remove medium containing the remaining complexes from the cells by gentle aspiration, and wash cells once with 4 ml PBS. 10. Add fresh cell growth medium (containing serum and antibiotics). Assay cells for expression of the transfected gene after an appropriate incubation time.")
53
Transformation과 Transfection
Transformation과 Transfection- 제일 큰 차이는 host cell이다. Transformation 같은 경우는 박테리아 셀 내에 외래 DNA를 주입하는 것이고, transfection은 동물 세포에 주입하는 것을 말한다. 동물세포에는 transformation이 아닌 transfection이란 용어를 사용하는 이유는 동물세포에서의 transformation은 세포를 continuous cell line로 변형시킨다는 의미를 이미 가지고 있기 때문이며, 처음에 동물세포에 이뤄진 외래 DNA주입 실험이 phage DNA 를 벡터로 사용해 이뤄졌기 때문에 바이러스 infection에서 어미를 따서 transfection이란 용어를 사용하게 되었다. Competent cell 의 제조방법 LB plate에 자란 DH5 alpha colony를 LB 배지 2 ml에 접종한 후 밤새 키웁니다. 위에서 키운 배양액 1 ml을 100 ml LB 배지가 담긴 플라스크에 넣고 O.D.600이 0.4가 될 때까지 37°C shaking incubator에서 키웁니다. 배양액을 50 ml conical tube에 넣고 얼음에 10분간 놓아둡니다. 4°C에서 4,500 rpm으로 10분간 원심분리합니다. 상층액을 완전히 제거한 후 미리 차게 해 둔 0.1 M filtered CaCl2 용액을 원래의 1/2 부피로 넣고 부유시킵니다. 얼음에 15분 놓아두었다가 다시 4°C에서 3,000∼4,000 rpm으로 10분간 원심분리합니다. 상층액을 버리고 처음의 1/10 부피의 filtered CaCl2 용액으로 부유시킵니다. 얼음에 30분 놓아두었다가 미리 차게 해둔 microfuge tube에 200 ul씩 분주하고, 사용할 때까지 deep freezer에 보관합니다.

54
Some Cellular Organelles

55
Compartments/organelles examined
Protein sequences sufficient for localization Vital stains Mitochondria Nuclei Secretory Pathway: Endoplasmic Reticulum Golgi Complex Endocytotic Pathway: Endosomes

56
Nucleus Transport through nuclear pore
signal = basic amino acid stretches example: P-P-K-K-K-R-K-V

57
Import of proteins into nucleus through nuclear pore

58
Nuclear Stain: Hoechst binds DNA

59
Mitochondria Transmembrane transport signal
Example: H2N-M-L-S-L-R-Q-S-I-R-F-F-K-P-A-A-T-R-T-L-C-S-S-R-Y-L-L

60
Protein being transported across mitochondrial membranes

61
Mitochondrial dye = MitoTracker Red
Diffuses through membranes Non-fluorescent until oxidized Accumulates in mitochondria and oxidized Mitotracker DNA
![]()
62
Cellular components of the secretory and endocytic pathways
nuclear envelope endoplasmic reticulum lysosome early endosome late Golgi apparatus cis Golgi network trans stack CYTOSOL plasma membrane

63
Endoplasmic Reticulum
Entry into E.R.: Transmembrane transport signal = hydrophobic amino acid stretches Example: H2N-M-M-S-F-V-S-L-L-V-G-I-L-F-W-A-T-E-A-E-Q-L-T-K-C-E-V-F-Q at amino terminus Retention in E.R. lumen: Signal = K-D-E-L-COOH at carboxy terminus

64
Endoplasmic Reticulum marker
ER-Tracker Blue-White Live bovine pulmonary artery endothelial cells

65
Mitotracker Red and ER-blue/white
![]()
66
Golgi nucleus From the ER, secreted and membrane proteins move to the Golgi, a series of membrane-bound compartments found near the nucleus

67
Golgi marker BODIPY-TR ceramide Ceramide = lipid
When metabolized, concentrates in the Golgi Red fluorophore

68
Cultured Epithelial Cells
DNA (Hoechst) Golgi (ceramide)
 Golgi (ceramide)")
69
MDCK Cells DNA (Hoechst) Golgi (ceramide) Lysosomes (LysoTracker)
Madin-Darby Canine Kidney Polarized Epithelial Cells DNA (Hoechst) Golgi (ceramide) Lysosomes (LysoTracker) Molecular Probes, Inc.
![]()
70
Rhodamine transferrin
Does the fluorescent green protein co-localize?

71
Immunofluorescence / confocal microscopy 원리

72
Immunofluorescence / confocal microscopy 실험방법
B or T cells in suspension, adherent cells on chambered coverglass or chamberslides, cryostat sections of unfixed, OCT embedded tissue: 1. wash cells 1x cold RPMI (no wash for cryostat sections). 2. Fix 20 min 4% paraformaldehyde in 0.1M phosphate buffer pH 7.4, 0.03M sucrose on ice. ** 3. wash 2x PBS/1%BSA. From now on everything can be at room temp. or on ice. 4. Permeabilize 5 min RT in 0.2% saponin, PBS, 0.03M sucrose, 1% BSA. 5. wash 1x PBS/BSA. 6. Block 15 min 5% normal goat serum (NGS) in PBS/BSA. 7. wash 1x PBS/BSA. 8. 1° diluted in PBS/BSA 60 min RT; 100 l per tube or section. 9. wash 3x PBS/BSA. 10. Block 15 min 5% NGS in PBS/BSA. 11. wash 1x PBS/BSA. 12. 2° diluted in PBS/BSA 30 min RT; 100 l per tube or section. 13. wash 3x PBS/BSA; (wash 1x in PBS/BSA then 2 x 10 min in Molecular Probes SlowFade Light buffer if using Slow Fade Light S-7461 to coversip); pellet cells and put up in two drops of Molecular Probes Slowfade Light antifade medium. Pipet about 15 m l on slide and coverslip. For chambered coverglass just put two-three drops in each chamber after wash.
. 2. Fix 20 min 4% paraformaldehyde in 0.1M phosphate buffer pH 7.4, 0.03M sucrose on ice. ** 3. wash 2x PBS/1%BSA. From now on everything can be at room temp. or on ice. 4. Permeabilize 5 min RT in 0.2% saponin, PBS, 0.03M sucrose, 1% BSA. 5. wash 1x PBS/BSA. 6. Block 15 min 5% normal goat serum (NGS) in PBS/BSA. 7. wash 1x PBS/BSA. 8. 1° diluted in PBS/BSA 60 min RT; 100 l per tube or section. 9. wash 3x PBS/BSA. 10. Block 15 min 5% NGS in PBS/BSA. 11. wash 1x PBS/BSA ° diluted in PBS/BSA 30 min RT; 100 l per tube or section. 13. wash 3x PBS/BSA; (wash 1x in PBS/BSA then 2 x 10 min in Molecular Probes SlowFade Light buffer if using Slow Fade Light S-7461 to coversip); pellet cells and put up in two drops of Molecular Probes Slowfade Light antifade medium. Pipet about 15 m l on slide and coverslip. For chambered coverglass just put two-three drops in each chamber after wash.")
Similar presentations






>")
 은 여러 단백질의 혼합물로부터 찾고자 하는 단백질 에 대한 항체를 사용하여 항원 - 항체 반응을 일으킴 으로써 특정단백질을 찾아내는.>")
 5주차 Subcloning Ⅱ : Detection of Subcloning - Rapid Microscale Isolation of Plasmid from Transformed Cells 담당교수 : 하상준 교수님 담당조교 : 조소영.>")

 전기영동 (Electrophoresis) 전하를 띤 고분자 물질을 전기장을 띤 매질 ( 젤 ) 에서 이동 ∙ 분리 물질의 분리, 순도 ∙ 특성 분석 물질의 이동.>")

. Why must organisms reproduce?>")




 6 주차 조교 : 전지선.>")


>")




